
Obsah článku
- Pfeifferův syndrom je genetická porucha charakterizovaná předčasným srůstem určitých kostí lebky (kraniosynostóza). Tento předčasný srůst zabraňuje normálnímu růstu lebky a ovlivňuje tvar hlavy a obličeje.
- Pfeifferův syndrom také ovlivňuje kosti v rukou a nohou.
- Mnoho charakteristických rysů obličeje Pfeifferova syndromu je výsledkem předčasného srůstu kostí lebky. Abnormální srůst těchto kostí vede k vypouleným a široce posazeným očím, vysokému čelu, nedostatečně vyvinuté horní čelisti a zobákovitému nosu. Více než polovina všech dětí s Pfeifferovým syndromem má ztrátu sluchu; časté jsou také problémy se zuby.
 Pfeifferův syndrom je vzácná genetická porucha charakterizovaná předčasným splynutím určitých kostí lebky (kraniosynostóza) a abnormálně širokými palci a prsty na nohou.
Pfeifferův syndrom je vzácná genetická porucha charakterizovaná předčasným splynutím určitých kostí lebky (kraniosynostóza) a abnormálně širokými palci a prsty na nohou.
Rozeznávají se tři formy Pfeifferova syndromu, z nichž nejzávažnější jsou typy II a III. Pfeifferův syndrom je autozomálně dominantní stav spojený s mutacemi v genech fibroblastový růstový faktor receptor 2 (FGFR2) a fibroblastový růstový faktor receptor 1 (FGFR1).
Nyní je známo, že Pfeifferův syndrom je členem skupiny stavů způsobených mutacemi v genech FGFR, včetně Apertova syndromu, Crouzonova syndromu, Beare-Stevensonova syndromu, izolované koronální synostózy související s FGFR2, Jackson-Weissova syndromu.
Příznaky Pfeifferova syndromu
Kojenci s Pfeifferovým syndromem typu I mají kraniosynostózu, která způsobuje, že je hlava krátká a vysoká. Mezi další příznaky může patřit vysoké čelo; nedostatečně rozvinuté oblasti střední části obličeje; široce rozmístěné oči (oční hypertelorismus); nedostatečně vyvinutá horní čelist (hypoplastická horní čelist) s výraznou dolní čelistí; a zubní abnormality. Inteligence je obvykle normální.
Kraniosynostóza je termín označující předčasný srůst švů lebky, vznikající v prvních týdnech a měsících po narození. Vzhledem k tomu, že mozek nadále roste rychlým tempem, najde si cestu nejmenšího odporu a nakonec dojde ke změně tvaru mozku a lebky. Každý předčasně uzavřený šev, vede k určitému abnormálnímu tvaru hlavy.
Pfeifferův syndrom typu II je charakterizován závažnější formou kraniosynostózy (lebka do tvaru čtyřlístku), se závažnějšími anomáliemi rukou a nohou a dalšími malformacemi končetin. U kojenců s Pfeifferovým syndromem typu II předčasné uzavření lebečních švů způsobí, že lebka bude mít „trojlaločný“ vzhled. Kromě toho je tato forma kraniosynostózy často spojena s hydrocefalem, stavem, při kterém je změněn normální tok mozkomíšního moku, což vede k abnormálnímu rozšíření (dilataci) prostorů v mozku (komorách), což způsobuje akumulaci mozkomíšního moku v lebce a vzniká zvýšený tlak na mozek. Charakteristické kraniofaciální rysy spojené s Pfeifferovým syndromem typu II mohou zahrnovat abnormálně vysoké a široké čelo; silně vypouklé oči (tj. oční proptóza); neobvykle plochá střední část obličeje; „zobákovitý“ nos; a dolů posunuté uši. Postižení kojenci mohou také vykazovat abnormální fixaci a nedostatečnou pohyblivost (ankylózu) loketních kloubů a / nebo v některých případech různé malformace určitých vnitřních orgánů v břiše (viscerální anomálie). U kojenců s Pfeifferovým syndromem typu II navíc často dochází k narušení duševního vývoje a neurologickým problémům v důsledku závažného postižení mozku a / nebo k hypoxii v důsledku problémů s dýcháním. Bez vhodné léčby mohou fyzické abnormality spojené s poruchou vést k život ohrožujícím komplikacím během kojeneckého věku.
Typ 2 se odlišuje od typu 3 přítomností hlavy ve tvaru čtyřlístku, která je způsobena rozsáhlejším spojením kostí v lebce.
Jedinci s Pfeifferovým syndromem typu III mají příznaky a nálezy podobné těm, které jsou přítomny u Pfeifferova syndromu typu II, s výjimkou deformace lebky do čtyřlístku. Mezi další charakteristiky spojené s Pfeifferovým syndromem typu III patří zkrácená základna lebky (přední); abnormální přítomnost určitých zubů při narození (natální zuby); silný vypouklé oči v důsledku abnormální povrchnosti kostních dutin, které se přizpůsobují očním bulvám; a / nebo různé malformace určitých vnitřních orgánů v břišní oblasti (viscerální anomálie). Stejně jako u typu II se u osob s Pfeifferovým syndromem typu III často vyskytuje zhoršený duševní vývoj a závažné neurologické problémy a mohou se u nich vyvinout potenciálně život ohrožující komplikace již v raném věku bez vhodné léčby.
Sponzorováno
Typy 2 a 3 jsou závažnější formy Pfeifferova syndromu, které často zahrnují problémy s nervovým systémem.
Příčiny Pfeifferova syndromu
Pfeifferův syndrom je autozomálně dominantní genetická porucha. Dominantní genetické poruchy se vyskytují, když je k vyvolání určité nemoci nezbytná pouze jedna kopie abnormálního genu. Abnormální gen může být zděděn od jednoho z rodičů nebo může být výsledkem nové mutace (změna genu) u postiženého jedince. V podstatě všechny případy Pfeifferova syndromu typu II a typu III vyplynuly z nových mutací.
Pokročilý věk otců je spojen se zvýšeným rizikem nových mutací pro Pfeifferův syndrom. Riziko přenosu abnormálního genu z postiženého rodiče na potomstvo je 50% pro každé těhotenství. Riziko je stejné pro muže i ženy.
Pfeifferův syndrom typu I je spojen s mutacemi FGFR1 a FGFR2. Pfeifferův syndrom typu II a typu III je spojen s mutacemi v FGFR2.
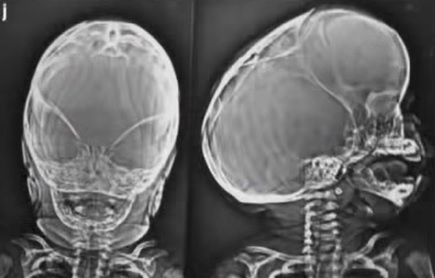
Pfeifferův syndrom – rentgen lebky
Léčba Pfeifferova syndromu
Léčba Pfeifferova syndromu je zaměřena na specifické příznaky, které jsou patrné u každého jedince. Léčba může vyžadovat koordinované úsilí týmu specialistů. Pediatři, chirurgové, lékaři, kteří diagnostikují a léčí poruchy uší, nosu a krku (otolaryngologové), neurologové, specialisté, kteří posuzují a léčí problémy se sluchem (audiologové), a / nebo další lékaři a specialisté možná budou muset systematicky a komplexně plánovat léčbu postiženého dítěte.
Specifické terapie pro Pfeifferův syndrom jsou symptomatické a podpůrné. Protože kraniosynostóza a v některých případech i přidružený hydrocefalus mohou mít za následek abnormálně zvýšený tlak v lebce (intrakraniální tlak) a v mozku, lze doporučit včasný chirurgický zákrok k nápravě kraniosynostózy a v případě hydrocefalu k zavedení trubice k odtoku přebytečné mozkomíšní tekutiny pryč z mozku a do jiné části těla, kde může být mozkomíšní mok absorbován. Včasnou korekční a rekonstrukční operaci lze provést také u kojenců s Pfeifferovým syndromem, aby se pomohlo napravit určité související kraniofaciální abnormality (např. problémy obličeje, asymetrie obličeje, abnormality nosu, očí).
Může dojít také k problémům dýchacích cest, zejména u velmi malých dětí. To způsobuje nízkou hladinu kyslíku, která, pokud není rozpoznána a léčena, může vést k poškození mozku. Takže na to pozor.
Kromě toho může být v některých případech provedena rekonstrukční chirurgie, která pomůže napravit malformace ucha, a / nebo mohou být použity speciální naslouchátka ke zlepšení sluchu.
U některých jedinců s Pfeifferovým syndromem lze provést chirurgický zákrok, který pomůže napravit syndaktylii a / nebo jiné malformace skeletu a zlepšit funkci a mobilitu. Fyzikální terapie a další ortopedická a podpůrná opatření mohou být také použita k dalšímu zlepšení mobility postiženého jedince. Chirurgické zákroky prováděné za účelem korekce určitých kraniofaciálních, audiologických a / nebo skeletálních abnormalit spojených s poruchou budou záviset na závažnosti a umístění anatomických abnormalit a jejich souvisejících symptomech.
Včasná intervence může být důležitá, aby se zajistilo, že děti s Pfeifferovým syndromem dosáhnou svého potenciálu. Mezi speciální služby, které mohou být prospěšné pro postižené děti, patří speciální sociální podpora, fyzioterapie a další lékařské, sociální a / nebo odborné služby.
Pfeifferův syndrom postihuje přibližně 1 ze 100 000 jedinců.
Sponzorováno
U postižených jedinců a jejich rodin se doporučuje genetické poradenství. Kromě toho mohou být u rodinných příslušníků diagnostikovaných jedinců důležitá důkladná klinická hodnocení, aby se zjistily jakékoli příznaky a fyzické vlastnosti, které mohou potenciálně souviset s Pfeifferovým syndromem.
A dál si přečtěte
Zdroj článku a studie na toto téma
- Studie - Případ Pfeifferova syndromu Autor: Moon Sung Park
- Pfeiffer syndrome: Clinical and genetic findings in five Brazilian families Autor: Hercílio-Martelli Júnior
- Studie o Pfeifferově syndromu Autor: Annick Vogels
Sponzorováno
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění, Vzácná onemocnění
Přečtěte si také naše další články