
Obsah článku
Mowat-Wilsonův syndrom
Mowat-Wilsonův syndrom je způsoben mutací genu ZEB2. Obvykle se vyskytuje poprvé u osoby s tímto syndromem a není zděděn od rodiče. Jen zřídka se objeví více než jedno dítě s MWS v rodině. Léčba závisí na přítomných příznacích a zaměřuje se na specifické potřeby každého člověka.
Známky a příznaky Mowat-Wilsonova syndromu
Mowat-Wilsonův syndrom je spojen s řadou fyzických příznaků i mentálním postižením. Většina lidí s MWS má těžké mentální postižení, ačkoli malý počet pacietů má mírnější rysy a jen mírné mentální postižení. Lidé, kteří mají Mowat-wilsonův syndrom, mají obvykle výrazný vzhled obličeje, chybí nebo mají silně omezenou řeč a často mívají záchvaty.
Některé fyzické problémy se mohou projevit při narození nebo v kojeneckém věku. Patří mezi ně střevní porucha Hirschsprungova choroba (asi u poloviny), problémy s vývojem ledvin a mužských genitálií (hypospadias), vrozené srdeční vady, problémy s očima a absence oblasti mozku, která spojuje obě mozkové hemisféry (ageneze corpus callosum). Pozdější problémy mohou zahrnovat malou velikost hlavy (mikrocefalie) a nízkou postavu. Chronická zácpa se může objevit i u těch pacientů, kteří nemají Hirschsprungovu chorobu.
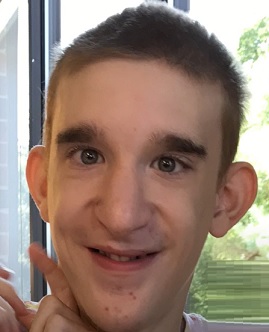
Výrazný vzhled obličeje lidí s MWS je nejkonzistentnějším rysem tohoto stavu a může ho rozpoznat zkušený lékařský specialista. Mezi společné rysy patří vysoké čelo, široké obočí, které jsou centrálně od sebe vzdálené, oči s velkým rozestupem (hypertelorismus), které jsou velké a hluboko posazené, povznesené ušní laloky s centrální prohlubní, relativně malý nos (u kojenců) s výraznou zaoblenou špičkou nosu, prominentní část mezi nosními dírkami (columella), otevřená ústa s horním rtem ve tvaru písmene M a prominentní, ale úzká a trojúhelníková špičatá brada. U kojenců nemusí být všechny tyto vlastnosti zřejmé. Některé rysy obličeje se časem stávají zřetelnějšími, takže diagnóza Mowat-Wilsonova syndromu je u starších dětí snadnější.
Děti s Mowat-Wilsonovým syndromem dosahují svého vývojového pokroku (jako je sezení a chůze) výrazně pomaleji, než je průměr. Řeč je často opožděná nebo chybí, až na několik výjimek (tj. mírný Mowat-Wilsonův syndrom). Porozumění je obvykle lepší než schopnost řeči a děti mohou komunikovat neverbálně, například podpisem nebo použitím komunikačního zařízení. Obvykle se chovají šťastně a často se usmívají.

Pacientka s Mowat-Wilsonovým syndromem
Záchvaty jsou běžné a vyskytují se přibližně u 90% jedinců ve věku 10 let. Záchvaty mohou být v dětství obtížně kontrolovatelné, ale v dospělosti obvykle nejsou velkým problémem. Malá podskupina jedinců bude mít během spánku Status epilepticus, který může vést ke ztrátě některých vývojových a fyzických dovedností, pokud nebude nadále léčen.
Ačkoli se u kojenců s MWS může objevit řada vrozených abnormalit, je důležité si uvědomit, že postižení kojenci nebudou mít všechny anomálie spojené se stavem. Jednou běžnou vrozenou abnormalitou je Hirschsprungova choroba, gastrointestinální stav charakterizovaný absencí určitých nervových buněk (ganglií) ve stěně hladkého svalstva v oblasti tlustého střeva. Výsledkem je absence nebo narušení nedobrovolných rytmických kontrakcí, které pohánějí potravu gastrointestinálním traktem (peristaltika). Mezi příznaky Hirschsprungovy choroby patří zácpa, zvracení, ztráta chuti k jídlu, nadýmání nebo otoky (distenze) břicha, abnormální hromadění výkalů v tlustém střevě a rozšíření tlustého střeva nad postiženým segmentem (megakolon). Hirschsprungova choroba může nakonec způsobit průjem, dehydrataci a selhání růstu a přibývání na váze očekávanou rychlostí (selhání prospívání). Nízký vzrůst je v MWS běžný, i když někteří lidé mají normální vzrůst.
Sponzorováno
Jedním vzácným nálezem je nepřítomnost sleziny, orgánu, který pomáhá v boji proti určitým typům infekcí. Všichni jedinci s MWS by měli být v době diagnózy vyšetřeni, zda mají slezinu. To lze provést pomocí ultrazvuku břicha. Krevní testy mohou někdy také naznačovat, že slezina chybí.
Mírný Mowat-Wilsonův syndrom
Byla identifikována podmnožina jedinců s MWS, kteří mají mírné rysy. Tito jedinci nemusí mít žádné malformace nebo mohou mít méně obličejových rysů, což někdy činí diagnostiku obtížnější. Navíc mohou mít takoví jedinci mentální postižení, které spadá do mírného, na rozdíl od těžkého rozsahu. Schopnosti řeči jsou pokročilejší a někteří pacienti jsou schopni mluvit v krátkých větách do poloviny dětství.
Příčiny Mowat-Wilsonova syndromu
MWS je autozomálně dominantní genetická porucha způsobená abnormalitou (mutací) v genu zvaném ZEB2. Tato mutace vede buď ke ztrátě funkce (běžné), nebo ke snížení funkce (vzácné) tohoto genu. Gen TheZEB2 (dříve nazývaný ZFHX1BorSIP1) se nachází na chromozomu 2 v oblasti 2q22.3. Geny poskytují pokyny pro výrobu proteinu, který hraje rozhodující roli při tvorbě mnoha orgánů a tkání v těle před narozením.
Když dojde k mutaci v jedné kopii tohoto genu, může být produkovaný protein chybný, neúčinný nebo chybí. To ovlivňuje vývoj mnoha orgánů a tkání v celém těle, zejména mozku. Mowat-Wilsonův syndrom se téměř vždy vyskytuje jako nová (sporadická nebo de novo) mutace. To znamená, že téměř ve všech případech došlo k genové mutaci v době tvorby vajíčka nebo spermií pouze pro toto dítě a žádný další člen rodiny nebude ovlivněn. Obvykle není syndrom zděděn od zdravého rodiče.
Ve velmi malém počtu rodin bylo MWS postiženo více než jedno dítě. Z více než 300 jedinců popsaných v literatuře byl recidiva MWS hlášena pouze u 5 rodin.
Diagnóza Mowat-Wilsonova syndromu
Stanovení diagnózy může být často náročné. Lékaři se při stanovení diagnózy obvykle dívají na anamnézu, příznaky, fyzikální vyšetření a výsledky laboratorních testů.
Mowat-Wilsonův syndrom je obvykle diagnostikován v kojeneckém nebo dětském věku na základě důkladného klinického hodnocení, identifikace charakteristických fyzických nálezů a vzhledu obličeje a informací z různých specializovaných testů. Mnoho z těchto rysů se časem stává výraznějším, a tak je u starších jedinců snadnější stanovit diagnózu.
Kontrola funkcí může zahrnovat zobrazovací techniky, jako je skenování pomocí počítačové tomografie (CT) nebo zobrazování magnetickou rezonancí (MRI) mozku, ultrazvuk ledvin nebo ultrazvuk srdce.
Klinickou diagnózu lze potvrdit molekulárně genetickým testováním mutací v genu ZEB2.
Léčba Mowat-Wilsonova syndromu
Zacházení s jednotlivci pomocí Mowat-Wilsonovým syndromem by mělo být zaměřeno na potřeby každého jednotlivce. Možná bude nutné, aby tým specialistů spolupracoval a plánoval nejlepší strategii, která umožní každému jednotlivci plně využít svůj potenciál. Stejně jako každý jedinec bude jiný, bude plán léčby jedinečný a nejlépe projednaný se zdravotníky zapojenými do plánu péče.
V MWS vyžadují související stavy včetně Hirschsprungovy choroby, srdečních abnormalit a záchvatů zásah příslušných odborníků, jako jsou neurologové, kardiologové a chirurgové. Fyzikální terapie, pracovní terapie a logopedie mohou být užitečné při pomoci dětem s vývojovým zpožděním dosáhnout jejich plného potenciálu.
Léčba Hirschsprungovy choroby obvykle zahrnuje chirurgický zákrok k odstranění obstrukce střev. V břišní stěně se provede dočasné otevření tlustého střeva (kolostomie) a později se provede druhý chirurgický zákrok k odstranění nefunkční části tlustého střeva a opětovnému připojení ke zdravým částem střeva. Mohou být provedeny další operace k léčbě specifických vrozených anomálií, jako jsou srdeční vady a abnormality močových cest.
V některých případech byly záchvaty rezistentní na léčbu v dětství, ale zdá se, že jsou snáze zvládnutelné u dospívajících a dospělých.
U jedinců, kteří nemají funkční slezinu, lze zvážit podání specifických imunizací a někdy i denních antibiotik.
Sponzorováno
U postižených jedinců a jejich rodin se doporučuje genetické poradenství.
Zdroje článku a studie na toto téma
- Neuroimaging findings in Mowat-Wilson syndrome: a study of 54 patients Autor: Livia Garavelli, Ivan Ivanovski, Stefano Giuseppe Caraffi
- Mowat-Wilsonův syndrom: neurologická a molekulární studie u sedmi pacientů Autor: José Albino da Paz
Sponzorováno
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění, Vzácná onemocnění
Přečtěte si také naše další články