
Obsah článku
- Menkesova choroba, také známá jako Wilsonova nemoc výživy nebo Menkesův syndrom, je vzácné a genetické onemocnění, které postihuje metabolismus mědi v těle. Toto onemocnění je způsobeno mutacemi v genu ATP7A, který je odpovědný za přenos mědi v buňkách a její využití v různých enzymatických procesech.
- Důsledkem těchto genetických mutací je narušení metabolismu mědi, což má za následek nedostatečné vstřebávání mědi ze stravy a její neschopnost správně se dopravit do tkání a orgánů. To vede k abnormálnímu hromadění mědi v některých buňkách a nedostatku mědi v jiných, což má vážné důsledky pro funkci těla.
- Menkesova choroba je vážným onemocněním a neléčená může vést k trvalému poškození mozku a orgánů a může být fatální.
- Bohužel, neexistuje žádná účinná léčba, která by mohla úplně vyléčit Menkesovu chorobu. Nicméně, existují léčebné strategie, které mohou pomoci zmírnit některé z příznaků a zlepšit kvalitu života postižených jedinců.
- Léčba Menkesovy choroby vyžaduje multidisciplinární péči a specializované lékařské týmy, aby byla co nejefektivnější. Vzhledem k vzácnosti této choroby je důležité, aby byli pacienti s Menkesovou chorobou sledováni lékaři s odbornými znalostmi v tomto oboru, aby byla poskytnuta nejlepší možná péče a podpora.
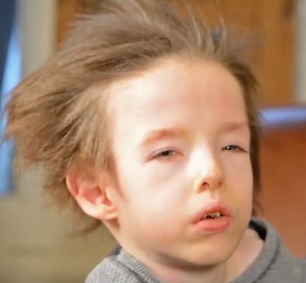 Menkesova choroba je genetická porucha metabolismu mědi, která je diagnostikovatelná už před narozením (prenatálně) a která znamená progresivně degenerativní ovlivnění několika orgánů v těle, zejména mozku. Vyznačuje se záchvaty, mentální retardací, zakrnělým růstem, celkovým neprospíváním, nestabilní tělesnou teplotou a velmi neobvyklou barvou a strukturou vlasů.
Menkesova choroba je genetická porucha metabolismu mědi, která je diagnostikovatelná už před narozením (prenatálně) a která znamená progresivně degenerativní ovlivnění několika orgánů v těle, zejména mozku. Vyznačuje se záchvaty, mentální retardací, zakrnělým růstem, celkovým neprospíváním, nestabilní tělesnou teplotou a velmi neobvyklou barvou a strukturou vlasů.
Příznaky poruchy jsou způsobeny selháním transportních systémů mědi uvnitř buňky přes buněčnou membránu. Kvůli selhání tohoto transportního systému je měď nedostupná pro různé buňky – měď je ale nezbytná pro strukturu a funkci různých enzymů, které řídí vývoj vlasů, mozku, kostí, jater a tepen.
Menkesova choroba je genetická porucha způsobená mutacemi v genu ATP7A, který je zodpovědný za produkci enzymu ATPázy, který reguluje hladiny mědi v těle.
Příznaky Menkesovy choroby
Menkesova choroba je charakterizována křehkými, řídkými, ocelově zbarvenými nebo zvrásněnými vlasy, které jsou často bílé, slonovinové nebo šedé barvy. Mezi neobvyklé rysy obličeje patří jakoby zavalité tváře a neobvyklé obočí. Postižené děti se často rodí předčasně. Může se u nich objevit nižší než normální tělesná teplota (podchlazení) a nadbytek bilirubinu v krvi (neboli hyperbilirubinémie) způsobující žlutý vzhled (žloutenka).
U starších kojenců se může objevit také podchlazení. V některých případech se tyto časné příznaky mohou ztratit a normální nebo mírně zpomalený vývoj může probíhat po dobu dvou až tří měsíců. Může dojít i k vážnému zpoždění vývoje, ke ztrátě raných, vývojových dovedností a ke křečím. Abnormality mozku, jako je krevní sraženina na mozku (subdurální hematom) a / nebo ruptura nebo trombóza tepen v mozku také nejsou úplně neobvyklé. Oslabené kosti (osteoporóza) jsou běžné a mohou vést snadno ke zlomeninám. Kombinace subdurálního hematomu a zlomenin kostí může vést k nesprávné diagnóze zneužívání dětí. Rovněž byl popsán emfyzém, abnormality močového měchýře, degenerace sítnice a cysty duhovky. Ve vzácných případech jsou příznaky velmi mírné a může se objevit jen několik typických příznaků.
Sponzorováno
Příčiny Menkesovy choroby
Menkesova choroba je na chromozom X vázaná choroba spojená s genetickým defektem ATP-ázy – tedy způsobená mutacemi v genu ATP7A. Tento gen je zodpovědný za produkci enzymu ATPázy, který reguluje hladiny mědi v těle. Jedinci s Menkesovou chorobou mají neobvykle nízkou hladinu mědi v mozku a játrech a přebytek mědi ve střevech a ledvinách. Bez mědi jako klíčového prvku selhávají přirozené enzymy závislé na mědi.
Experimentální důkazy jsou zřejmé, že bez vazby mědi na peptid aminokyseliny je příčinou podchlazení enzym cytochrom-c oxidáza; defektní enzym tyrosináza je příčinou nedostatku pigmentu na vlasech a kůži; defektní enzym lysyl oxidáza je příčinou selhání pojivové tkáně k vytvoření silných vnitřních stěn krevních cév; defektní enzym monoamin oxidáza je příčinou problémů vlasů; a defektní enzym askorbát oxidáza je příčinou nestandardní tvorby kosti.
Genové poruchy spojené s X jsou stavy způsobené abnormálním genem na chromozomu X a vyskytují se většinou u mužů. Ženy, které mají chorobný gen přítomný na jednom ze svých X chromozomů, jsou nositeli této poruchy. Tyto ženy obvykle nevykazují příznaky, protože ženy mají dva chromozomy X a jeden je inaktivován, takže geny na tomto chromozomu nefungují. Muži mají jeden chromozom X, který je zděděn od své matky, a pokud muž zdědí chromozom X, který obsahuje gen choroby, bude se nemoc rozvíjet.
Nedávné studie naznačují, že výskyt Menkesovy choroby se pohybuje od přibližně 1 z 100 000 narozených dětí do 1 z 250 000 narozených dětí.
Související poruchy
Příznaky následujících poruch mohou být podobné příznakům Menkesovy choroby. Srovnání může být užitečné pro diferenciální diagnostiku:
Wilsonova nemoc je vzácná genetická porucha metabolismu mědi charakterizovaná nadměrným ukládáním mědi v tělních tkáních, zejména v játrech, mozku a rohovkách očí. To vede nakonec k onemocnění jater, dysfunkci mozku a charakteristickému rezavě hnědému zbarvení prstence kolem rohovky každého oka známého jako Kayser-Fleischerův prstenec. Symptomy se obvykle neobjeví v časném dětství.
Primární biliární cirhóza, známá také jako Hanotova cirhóza, je vzácný stav vyskytující se hlavně u žen.
Indická dětská cirhóza je familiární a pravděpodobně geneticky podmíněná nemoc. V játrech se hromadí extrémně velké množství mědi, což je fenomén podobný Menkesově chorobě.
Diagnostika Menkesovy choroby
Menkesova choroba je diagnostikována měřením sníženého množství mědi a ceruloplasminu v krevní plazmě, ale tyto testy nejsou vždy spolehlivé (zejména v novorozeneckém období). Nová metoda diagnostiky, která může potenciálně identifikovat postižené děti dříve, než nedostatek mědi ovlivní mozek, zahrnuje měření hladin katecholaminů v krevní plazmě. K potvrzení diagnózy je k dispozici molekulární genetické testování mutací v genu APT7A. Testování nosiče a prenatální diagnostika jsou také k dispozici, pokud je u postiženého člena rodiny mutace ATP7A.
Léčba Menkesovy choroby
Sponzorováno
Prenatální nebo velmi časná (do týdnů) diagnóza Menkesovy choroby je nezbytná. Ukázalo se, že injekce sloučeniny histidinu a mědi zvyšují koncentraci mědi v krvi a jsou tak dostupné pro vývoj enzymů závislých na mědi a pro produkci myelinu. Účinnost léčby histidinu a mědi závisí na tom, zda byla zachována určitá aktivita enzymu APT7A a zda bylo rychle zahájeno doplňování mědi.
Studie a zdroje článku
- Menkes Kinky Hair Disease Autoři: Praveen Kumar Ramani; Bindu Parayil Sankaran.
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění, Vzácná onemocnění
Přečtěte si také naše další články