
Obsah článku
- Apertův syndrom je vzácná vrozená genetická porucha, která se vyznačuje předčasným srůstem některých lebečních švů, což vede k abnormalitám ve tvaru hlavy a obličeje. Tento stav je známý jako kraniosynostóza.
- Apertův syndrom také způsobuje srůst prstů na rukou a nohou (syndaktylie) a může mít vliv na vývoj mozku a inteligenci.
- Apertův syndrom je způsoben mutací v genu FGFR2 (receptor pro fibroblastový růstový faktor), který hraje roli ve vývoji kostí. Tato genetická mutace je obvykle náhodná, ale může být i dědičná (v případě autosomálně dominantního vzoru dědičnosti).
- Diagnóza Apertova syndromu se obvykle provádí na základě klinických příznaků a fyzického vyšetření novorozence. K potvrzení může být použito genetické testování, které identifikuje mutaci v genu FGFR2.
 Apertův syndrom je vzácný genetický stav, který je zjevný při narození. Děti s Apertovým syndromem mohou mít výrazné malformace lebky, obličeje, rukou a nohou. Apertův syndrom je charakterizován kraniosynostózou, stavem, při kterém se lebeční švy mezi kostmi lebky předčasně uzavírají. To může způsobit, že horní část hlavy vypadá špičatě a může to ovlivnit i kosti obličeje.
Apertův syndrom je vzácný genetický stav, který je zjevný při narození. Děti s Apertovým syndromem mohou mít výrazné malformace lebky, obličeje, rukou a nohou. Apertův syndrom je charakterizován kraniosynostózou, stavem, při kterém se lebeční švy mezi kostmi lebky předčasně uzavírají. To může způsobit, že horní část hlavy vypadá špičatě a může to ovlivnit i kosti obličeje.
Některé prsty na rukou nebo nohou mohou být spojené dohromady. Postižené děti mohou mít také mentální postižení. Závažnost příznaků se u jednotlivců liší. Apertův syndrom téměř vždy vzniká z nových genetických změn (mutací), ke kterým dochází náhodně. Zřídka se dědí autosomálně dominantním vzorem. Lidé s Apertovým syndromem mohou podstoupit různé terapie zaměřené na specifické příznaky. To může zahrnovat rekonstrukční operace lebky, obličeje, rukou a nohou.
Příznaky Apertova syndromu
Apertův syndrom je charakterizován kraniosynostózou, předčasným uzavřením lebečních švů mezi určitými kostmi v lebce. U jedinců bez kraniosynostózy umožňují lebeční švy růst a dovolují tak expanzi hlavy dítěte. Nakonec se tyto kosti spojí a vytvoří lebku. U dětí s kraniosynostózou mozek stále roste i po předčasném uzavření těchto švů. Tlak růstu mozku může způsobit, že různé kosti lebky a obličeje se během vývoje změní. V závislosti na tom, které lebeční švy se předčasně uzavírají, se jejich závažnost může lišit. U většiny postižených jedinců dochází k předčasnému uzavření lebečních švů mezi kostmi tvořícími čelo a horní strany lebky. To má pak tendenci způsobovat, že se hlava od narození jeví špičatá (akrocefalie). Kromě toho může zadní část lebky vypadat zploštělá s vysokým a širokým čelem. Jednotlivci mohou mít také hydrocefalus, při kterém se mozkomíšní mok abnormálně hromadí v dutinách mozku. To pak může způsobit tlak na mozek.
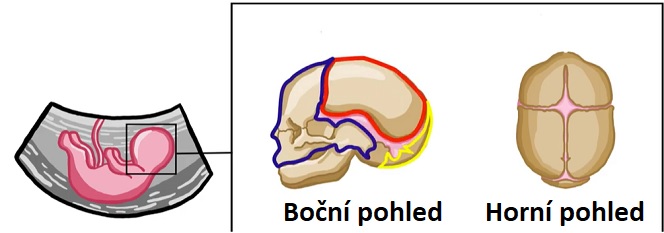
Apertův syndrom je charakterizován kraniosynostózou
Kosti obličeje tedy mohou být ovlivněny kraniosynostózou. To může vést k charakteristickým abnormalitám obličeje. Lidé s Apertovým syndromem mohou mít oči do široka od sebe (hypertelorismus), vypouklé oči nebo šikmé oční štěrbiny. Mohou mít také nedostatečně rozvinuté oblasti středního obličeje (maxilární hypoplázie) a abnormality patra, jako je rozštěp patra. Pravá a levá strana obličeje nemusí být symetrická. Lidé s Apertovým syndromem mohou mít zploštělý nos. Jednotlivci mohou mít zpožděný růst zubů, shlukování zubů nebo otevřený skus. Mohou mít středně silné až silné akné. Pokud jsou otvory mezi nosem a hrdlem zúžené nebo ucpané nebo je tracheální chrupavka deformována, mohlo by to narušit dýchání a polykání. Jedinci s těmito blokádami mohou mít infekce horních cest dýchacích, spánkovou apnoe a pravděpodobně i podvýživu.
Apertův syndrom má několik charakteristických malformací rukou a nohou. Postižení jedinci mohou mít krátké prsty a široké palce a prsty na nohou, které se odchylují směrem ven. Mohou také mít částečnou až úplnou fúzi (tj. syndaktylie) určitých prstů na rukou a nohou. Mnoho postižených lidí má úplnou fúzi (spojení) kostí druhého až čtvrtého prstu a jeden palec (podobně jako rukavice palčáky). Mohou však nastat i jiné fúze. Klouby prstů mají tendenci ztuhnout přibližně ve čtyřech letech. Na nohách spojení prstů také obvykle zasahuje druhý, třetí a čtvrtý prst. Nehty na nohou mohou být částečně spojené nebo oddělené. Apertův syndrom postihuje horní končetiny obecně více a závažněji než dolní končetiny.
Apertův syndrom může ovlivnit i jiné orgánové systémy:
Sponzorováno
Kosterní
- Snížení rychlosti růstu vedoucí k nízkému vzrůstu, navzdory normální porodní hmotnosti a porodní délce
- Srůst obratlů krku
- Srůst dvou kostí paží
- Srůst kostí zápěstí
Neurologické
- Různé stupně vývojového zpoždění
- Mírné až střední mentální postižení: Zdá se, že IQ závisí na faktorech, včetně věku operace dekomprese lebky a přítomnosti dalších mozkových anomálií.
- Absence corpus callosum, vláknité tkáně, která spojuje mozkové hemisféry mozku
- Neschopnost vytvořit membrány, které obvykle oddělují mozkové dutiny
- Zvětšená mozková dutina
- Malformace částí mozku, které se zabývají autonomním nervovým systémem (ANS). ANS řídí automatické funkce těla, jako je dýchání nebo srdeční frekvence
Uši
- Ztráta sluchu
- Chronické ušní infekce
Srdce (srdeční problémy)
- Otvory ve stěně komory
Břicho
- Užší otvor mezi dolní částí žaludku a horní částí tenkého střeva
- Blokování jícnu
Ledviny a urogenitální problémy
- Řitní otvor je mimo pozici
- Zablokování pochvy
- Selhání sestupu varlat
- Zvětšené ledviny v důsledku zablokování
Příčiny Apertova syndromu
Apertův syndrom je způsoben změnou (mutací) genu pro receptor pro fibroblastový růstový faktor 2 (FGFR2). Tento gen hraje zásadní roli ve vývoji kostry. Geny poskytují pokyny pro tvorbu proteinů, které hrají v našem těle odlišné role. Když dojde k mutaci genu, protein nemusí fungovat tak, jak by měl. U Apertova syndromu vedou mutace v FGFR2 k tomu, že tyto receptory nesprávně komunikují s růstovými faktory fibroblastů. To ovlivňuje tvorbu normálních lebečních švů a může to také bránit rozvoji mnoha dalších struktur v těle. To pak způsobuje malformace pozorované u Apertova syndromu.
U téměř všech hlášených pacientů byla porucha způsobena jednou ze dvou specifických mutací genu FGFR2. Tyto mutace jsou označeny jako „Ser252Trp“ a „Pro253Arg“. Tyto mutace mohou způsobit mírně odlišné prezentace, včetně závažnosti syndaktylie. Různé mutace v genu FGFR2 mohou způsobit několik dalších souvisejících poruch, včetně Pfeifferova syndromu, Crouzonova syndromu a Jackson-Weissova syndromu.
Až u 95% pacientů je Apertův syndrom výsledkem nové mutace genu FGFR2. Zdá se, že tyto nové mutace se vyskytují náhodně z neznámých důvodů (sporadicky). Bylo hlášeno, že sporadické případy mohou být spojeny se zvýšeným věkem otce.
Apertův syndrom se velmi zřídka dědí autozomálně dominantním způsobem. Dominantní genetické poruchy se vyskytují, když je k vyvolání určité nemoci nezbytná pouze jedna kopie mutace. Riziko přenosu mutace z postiženého rodiče na potomka je 50% pro každé těhotenství. Riziko je stejné pro muže i ženy.
Diagnostika
Apertův syndrom se diagnostikuje nejčastěji hned při narození, nebo pak v kojeneckém věku. Jedinec je diagnostikován prostřednictvím klinického hodnocení a řady specializovaných testů. Byly by identifikovány typické fyzické rysy, jako jsou anomálie obličeje nebo syndaktylie. Abnormality skeletu a vrozené srdeční vady lze detekovat pomocí zobrazovacích metod, jako je počítačová tomografie (CT) nebo MRI. Během screeningu sluchu u novorozence může být také detekováno poškození sluchu. Jednotlivci mohou také podstoupit testování mutací v genu FGFR2, které mohou poskytnout genetickou diagnózu Apertova syndromu.
V některých případech mohou být rysy Apertova syndromu detekovány i před narozením. To bývá provedeno pomocí prenatálního 2D nebo 3D ultrazvuku nebo magnetické rezonance (MRI). Ultrazvuk je neinvazivní procedura, která umožňuje vidět obraz plodu. To pak může detekovat rozdíly ve tvaru lebky, anomálie obličeje a syndactyly. MRI může poskytnout více podrobností o mozku plodu než ultrazvuk.
Léčba Apertova syndromu
Léčba Apertova syndromu se liší podle toho, jaké příznaky jsou u jednotlivce pozorovány. Taková léčba může vyžadovat péči týmu lékařů a specialistů, včetně pediatrů a chirurgů. Mezi specialisty mohou patřit specialisté na sluch, neurochirurgové, lékaři se specializací na poruchy kostí, kloubů a svalů (ortopedové), lékaři se specializací na poruchy uší, nosu a krku (otolaryngologové) a lékaři se specializací na srdeční abnormality (kardiologové).
Specifické terapie pro Apertův syndrom jsou symptomatické a podpůrné. Kraniosynostóza a hydrocefalus mohou mít za následek abnormálně zvýšený tlak v lebce a v mozku. V takových případech lze doporučit brzkou operaci (do 2 až 4 měsíců po narození) k nápravě kraniosynostózy. U pacientů s hydrocefalem může chirurgický zákrok zahrnovat také zavedení speciální trubice, která pak odvádí přebytečnou mozkomíšní tekutinu z mozku. Mozkomíšní mok může být odváděn do jiné části těla, kde by mohl být absorbován.
Může být doporučena korekční a rekonstrukční chirurgie, která pomůže napravit kraniofaciální malformace. Chirurgie může také pomoci napravit polydaktylii a syndaktylii a jiné kostní vady nebo fyzické abnormality. U osob s vrozenými srdečními vadami může být nutná léčba určitými léky, chirurgický zákrok a / nebo jiná opatření. U některých jedinců se sluchovým postižením mohou být třeba hodně prospěšná naslouchátka.
Včasná intervence může být důležitá, aby se zajistilo, že děti s Apertovým syndromem plně využijí svůj potenciál. Přínosem mohou být speciální služby, jako je fyzioterapie, pracovní terapie a speciální vzdělávání.
Sponzorováno
U postižených jedinců a jejich rodin se doporučuje genetické poradenství. Genetický poradce může vysvětlit příčiny Apertova syndromu. Může s vámi také diskutovat o rizicích, že budete mít další děti s Apertovým syndromem. Zásadní je také psychosociální podpora celé rodiny.
Zdroje článku a studie o Apertově syndromu
- Studie - Research advances in Apert syndrome Autoři: Satrupa Dasa a Anjana Munshic
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění
Přečtěte si také naše další články