
Obsah článku
- Adamsův-Oliverův syndrom je vzácné genetické onemocnění, které je charakterizováno kombinací kožních, kraniofaciálních a končetinových malformací. Tento syndrom je pojmenován po lékařích Adamsovi a Oliverovi, kteří jej poprvé popsali v roce 1945.
- Adamsův-Oliverův syndrom je geneticky heterogenní, což znamená, že jej mohou způsobovat mutace v různých genech. Bylo identifikováno několik genů spojených s tímto syndromem, včetně ARHGAP31, DOCK6, EOGT a RBPJ.
- Dědičnost může být autozomálně dominantní nebo autozomálně recesivní, v závislosti na konkrétní genetické mutaci.
- Diagnostika je založena na klinickém vyšetření a identifikaci charakteristických znaků. Genetické testy mohou potvrdit přítomnost specifických mutací.
- Neexistuje žádná specifická léčba pro Adamsův-Oliverův syndrom, ale léčba je zaměřena na zvládání jednotlivých příznaků a komplikací.
Adams-Oliverův syndrom (AOS) je extrémně vzácná dědičná porucha charakterizovaná vadami pokožky hlavy a abnormalitami prstů na rukou, nohou, pažích a / nebo nohou. Fyzické abnormality spojené s touto poruchou se u postižených jedinců velmi liší. Některé případy mohou být velmi mírné, zatímco jiné mohou být závažné. U kojenců s Adams-Oliverovým syndromem jsou vady pokožky hlavy přítomny při narození (tedy jsou vrozené) a mohou zahrnovat jednu nebo více zjizvených oblastí bez vlasů, které mohou mít abnormálně široké (rozšířené) cévy přímo pod postiženou kůží. V závažných případech může být přítomna i nějaká porucha kostí lebky. Navíc kojenci s touto poruchou mají obvykle malformace rukou, paží, nohou a / nebo nohou. Ty se pohybují od abnormálně krátkých (hypoplastických) prstů až po chybějící ruce a / nebo dolní končetiny. V některých případech mohou být přítomny i další abnormality.
Adamsův-Oliverův syndrom a jeho příznaky
Fyzické abnormality spojené s Adams-Oliverovým syndromem se u postižených jedinců velmi liší. Zatímco některé případy mohou být velmi mírné, jiné mohou být závažné. Nejčastějšími nálezy jsou vady končetin a pokožky hlavy.
Kojenci s Adams-Oliverovým syndromem mohou mít na pokožce hlavy jednu nebo více bezsrstých zjizvených oblastí kůže. Hloubka a velikost (průměr) lézí se velmi liší případ od případu. U některých postižených jedinců může dojít k ulceraci nebo infikování lézí na pokožce hlavy. Ve většině případů se však tyto kožní léze léčí i bez léčby (spontánně) během prvních několika měsíců života. U některých kojenců s Adams-Oliverovým syndromem mohou být pod kožními lézemi patrné abnormálně široké (rozšířené) krevní cévy. V závažných případech mohou tyto křehké rozšířené krevní cévy krvácet.
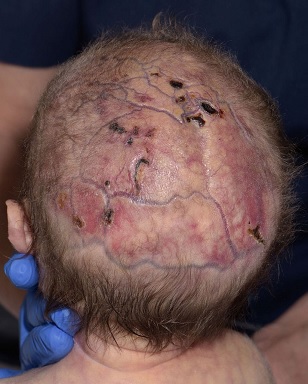
Takto mohou vypadat léze na hlavě u pacientů – Adamsův-Oliverův syndrom
V závažných případech mohou být defekty popsané výše spojeny se základním defektem lebky, včetně absence kosti v určitých oblastech lebky. Ve velmi závažných případech může dojít k odhalení určitých tkání pod lebkou, což má za následek zvýšenou náchylnost k nadměrnému krvácení a / nebo bakteriálním infekcím, jako je meningitida. Poruchy pokožky hlavy spojené s Adams-Oliverovým syndromem připomínají aplasia cutis congenita.
Děti s Adams-Oliverovým syndromem také vykazují malformace prstů na rukou, nohou, rukou a / nebo nohou. Někteří postižení kojenci mohou mít abnormálně krátké prsty na rukou a / nebo prstech na nohou v důsledku nedostatečného vývoje (hypoplázie) nebo absence určitých kostí v rukou a / nebo nohou. V závažných případech mohou prsty na nohou, ruce, chodidla a / nebo dolní končetiny částečně nebo zcela chybět. Dotčené osoby mohou mít také popruhy prstů na nohou (syndactyly) a nedostatečně vyvinuté (hypoplastické) nehty na nohou. Obecně bývají dolní končetiny (tj. dolní končetiny, chodidla a prsty na nohou) postiženy závažněji.
Adamsův-Oliverův syndrom a jeho příčiny
Zdá se, že většina případů AOS následuje autozomálně dominantní dědičnost. Dominantní genetické poruchy se vyskytují, když je k vyvolání určité nemoci nezbytná pouze jedna kopie abnormálního genu. Abnormální gen může být zděděn od jednoho z rodičů nebo může být výsledkem nové mutace (změna genu) u postiženého jedince. Riziko přenosu abnormálního genu z postiženého rodiče na potomstvo je 50% pro každé těhotenství. Riziko je stejné pro muže i ženy.
Sponzorováno
Fyzické nálezy spojené s Adams-Oliverovým syndromem (např. vady pokožky hlavy a abnormality prstů na rukou, nohou, pažích a / nebo nohou) se mohou od případu k případu velmi lišit (variabilní expresivita). Kromě toho se všechny vlastnosti spojené s touto poruchou nemusí projevit u všech, kteří gen zdědí.
Rovněž byla hlášena autozomálně recesivní dědičnost. Recesivní genetické poruchy se vyskytují, když jedinec zdědí dvě kopie abnormálního genu pro stejnou vlastnost, jednu od každého rodiče. Pokud jedinec obdrží jeden normální gen a jeden gen pro tuto nemoc, bude osoba nositelem této nemoci, ale obvykle nebude vykazovat příznaky. Riziko, že dva rodiče, kteří přenášejí, budou mít vadný genem a budou mít postižené dítě, je 25% s každým těhotenstvím. Riziko toho mít dítě, které je nosičem jako jeho rodiče, je 50% s každým těhotenstvím. Šance na to, aby dítě dostalo normální geny od obou rodičů a bylo geneticky normální pro tuto konkrétní vlastnost, je 25%. Riziko je stejné pro muže i ženy.
Někteří vědci se domnívají, že fyzické vlastnosti spojené s Adams-Oliverovým syndromem mohou být výsledkem přerušeného průtoku krve určitými tepnami během vývoje plodu. Pojem „Sekundární porucha přerušení zásobování tepny (SASDS)“ byl navržen pro skupinu vývojových poruch, ke kterým může dojít v důsledku narušení průtoku krve určitými tepnami během vývoj plodu. Mezi tyto poruchy patří Polandův syndrom (Poland syndrom), Klippel-Feilův syndrom, Moebiusův syndrom a Sprengelova deformita.
Kdo má často tento syndrom?
Adams-Oliverův syndrom je extrémně vzácná porucha, která podle všeho postihuje muže i ženy ve stejném počtu. V lékařské literatuře bylo popsáno více než 125 postižených jedinců. Hlavní fyzické rysy Adams-Oliverova syndromu (tj. poruchy pokožky hlavy a abnormality končetin) jsou zjevné při narození (vrozené).
Diagnóza
Diagnóza Adams-Oliverova syndromu může být podezřelá při narození na základě identifikace charakteristických defektů pokožky hlavy a lebky vyskytujících se v souvislosti s malformacemi prstů na rukou, nohou, rukou a / nebo nohou. Diagnóza může být potvrzena důkladným klinickým hodnocením, podrobnou anamnézou pacienta a řadou specializovaných testů, jako jsou pokročilé zobrazovací techniky. V některých případech je možné detekovat vady lebky a / nebo končetin před narozením (prenatálně) pomocí pokročilých zobrazovacích technik, jako je ultrazvuk. Během fetální ultrasonografie se odražené zvukové vlny používají k vytvoření obrazu vyvíjejícího se plodu.
Standardní terapie a léčba
Léčba Adams-Oliverova syndromu je zaměřena na specifické příznaky, které jsou patrné u každého jedince. Léčba může vyžadovat koordinované úsilí týmu specialistů. Je možné, že pediatři, ortopedičtí a plastičtí chirurgové, kardiologové, fyzioterapeuti a další zdravotničtí pracovníci budou muset systematicky a komplexně plánovat léčbu postiženého dítěte.
V mnoha případech se mohou vady pokožky hlavy léčit i bez léčby (spontánně) během několika prvních měsíců života. U postižených jedinců, kteří vykazují základní vady kostí lebky, může být nutné provést štěp kůže, kraniální chirurgii a / nebo jiné chirurgické zákroky. Lékaři mohou navíc doporučit, aby některé postižené děti nosily přilby, aby se zabránilo traumatu hlavy a možnému poškození abnormálně širokých (rozšířených) cév.
Fyzikální terapie, chirurgický zákrok a / nebo použití umělých končetin (protéz) lze doporučit dětem, které vykazují částečnou nebo úplnou absenci prstů na rukou, nohou, rukou, nohou a / nebo dolních končetin.
Postižené děti by navíc měly podstoupit kompletní lékařské vyšetření srdečních (srdečních) abnormalit, které jsou někdy spojeny s Adams-Oliverovým syndromem.
Sponzorováno
Genetické poradenství bude určitě přínosem pro postižené jedince a jejich rodiny.
Zdroj článku a studie
- Případ Adams-Oliverova syndromu - studie Autor: Minoo Saeidi
- Adams FH, Oliver CP. Hereditary deformities in man: Due to arrested development. J Hered. 1945;36:3–7
Sponzorováno
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění
Přečtěte si také naše další články