
Obsah článku
- Familiární adenomatózní polypóza je dědičné onemocnění, při kterém se v tlustém střevě a konečníku tvoří stovky až tisíce polypů (nezhoubné výrůstky sliznice).
- Familiární adenomatózní polypóza vzniká na základě mutace v genu APC (adenomatous polyposis coli), což zvyšuje riziko rozvoje rakoviny tlustého střeva.
- Pokud se polypy neléčí, téměř vždy se z některých z nich časem vyvine zhoubný nádor.
- V některých případech je nutné chirurgické odstranění části nebo celého tlustého střeva, aby se předešlo rakovině.
Co je familiární adenomatózní polypóza?
Familiární adenomatózní polypóza je genetická porucha, která vás předurčuje k rozvoji prekancerózních polypů tlustého střeva nazývaných adenomy. Polypy tlustého střeva jsou abnormální výrůstky ve výstelce tlustého střeva nebo konečníku. Nejsou to rakovina, ale některé typy, jako jsou právě adenomy, se mohou změnit na kolorektální rakovinu.
Mnoho lidí má několik polypů tlustého střeva náhodně, když stárnou. To nebývá zase až takový problém. Ale pokud máte dědičný polypózní syndrom, jako je familiární adenomatózní polypóza, vyvine se u vás mnoho polypů tlustého střeva – obvykle přes sto – počínaje mladým věkem. To výrazně zvyšuje vaše celoživotní riziko, že se jeden z nich stane rakovinným.
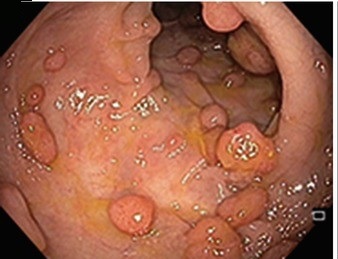
Náhled na polypy ve střevě
Ke zvládnutí tohoto rizika lékaři obvykle doporučují úplné odstranění tlustého střeva (totální kolektomie) a někdy i konečníku (to je pak proktokolektomie). Polypy se u familiární adenomatózní polypózy objevují až příliš často, než aby je bylo možné léčit a řešit jeden po druhém. Bez operace se u většiny lidí s familiární adenomatózní polypózou rozvine rakovina do jejich středního věku.
U lidí s familiární adenomatózní polypózou se také mohou vyvinout polypy v jiných orgánech a další abnormální výrůstky na místech, jako je kůže, měkké tkáně, zuby a kosti. I po operaci kolektomie budou muset pacienti pokračovat v pravidelných vyšetřeních na jiné nádory a možná v dalších operacích ke zvládnutí těchto nádorů.
Jaké je odhadované riziko rakoviny u familiární adenomatózní polypózy?
Bez léčby se riziko vzniku kolorektálního karcinomu s familiární adenomatózní polypózou blíží 100 %. S familiární adenomatózní polypózou se také vyvíjí relativně dříve a rychleji než u těch bez. Děti v rodinách, o nichž je známo, že jsou tímto syndromem postiženy, zahajují každoroční kolonoskopické screeningy ve věku kolem 10 let.
Kromě kolorektálního karcinomu mají lidé s familiární adenomatózní polypózou také zvýšené riziko vzniku dalších druhů rakoviny, jako třeba:
- Rakovina dvanáctníku (8 %)
- Papilární karcinom štítné žlázy (2 %)
- Rakovina slinivky břišní (2 %)
- Hepatoblastom (1,5 %)
- Rakovina žaludku (1 %)
- Nádory mozku a páteře (méně než 1 %)
Existují různé typy FAP?
Familiární adenomatózní polypóza má klasickou formu a některé méně běžné formy, které jsou považovány za podtypy.
- „Klasická“ familiární adenomatózní polypóza je charakterizována více než 100 adenomatózními polypy ve vašem tlustém střevě.
- „Atenuovaná“ familiární adenomatózní polypóza (AFAP) je méně závažný podtyp, ve kterém máte někde mezi 20 a 100 polypů v tlustém střevě.
- Gardnerův syndrom, stejně jako klasická familiární adenomatózní polypóza, typicky zahrnuje více než sto polypů tlustého střeva, stejně jako další typy nádorů v jiných částech těla.
- Turcotův syndrom zanamená mnohočetné polypy tlustého střeva, stejně jako jeden rakovinový nádor mozku.
Jak častá je familiární adenomatózní polypóza?
Familiární adenomatózní polypóza je vzácná, odhaduje se, že postihuje asi 1 z 8 000 lidí. Tvoří pouze asi 0,5 % všech případů kolorektálního karcinomu. Varianty jako AFAP, Gardnerův syndrom a Turcotův syndrom jsou ještě vzácnější. Představují mezi 5 % a 10 % všech případů familiární adenomatózní polypózy.
Jaký je rozdíl mezi familiární adenomatózní polypózou a Lynchovým syndromem?
Lynchův syndrom je jiný dědičný syndrom, který může zvýšit vaše riziko vzniku kolorektálního karcinomu a dalších druhů rakoviny. Lynchův syndrom nemusí nutně způsobit mnoho polypů tlustého střeva.
U lidí s Lynchovým syndromem se může vyvinout kolorektální karcinom s pouze jedním nebo několika polypy tlustého střeva. Polypy a rakovina mají také tendenci se vyvíjet o něco později než u klasické familiární adenomatózní polypózy a celoživotní riziko je o něco nižší. Různé genové mutace vedou k rozdílům mezi Lynchovým syndromem a familiární adenomatózní polypózou.
Sponzorováno
Jaké jsou příznaky familiární adenomatózní polypózy?
Polypy tlustého střeva u familiární adenomatózní polypózy se začínají objevovat mnohem dříve než u běžné populace, často v období dospívání. Obvykle polypy nezpůsobí příznaky, dokud nevyrostou dostatečně velké, aby byly nebezpečné. Ale pokud podstoupíte kolonoskopii pro účely screeningu, lékař je najde.
Lidé s familiární adenomatózní polypózou mají často stovky nebo tisíce polypů tlustého střeva. Ti s AFAP jich mají alespoň 20. Větší počet a rychlejší růst polypů u familiární adenomatózní polypózy může zvýšit pravděpodobnost, že způsobí příznaky. Příznaky mohou znamenat rektální krvácení, průjem nebo chronickou bolest břicha.
Lidé s familiární adenomatózní polypózou mohou mít také povrchové léze, které mohou lékaři pozorovat, jako například:
- Dermatofibromy, vláknité, jizvovité hrudky těsně pod kůží
- Epidermální cysty, kupolovité hrudky vyplněné keratinem těsně pod kůží
- Osteomy, nezhoubné kostní nádory, často viditelné na čelisti nebo lebce
- Více zubů nebo nevyrostlé zuby (viditelné na zubním rentgenu)
- Pigmentové léze na sítnici oka (vrozená hypertrofie retinálního pigmentového epitelu), viditelné při očním vyšetření. Obvykle nezpůsobují problémy se zrakem.
Jaký je průměrný věk nástupu příznaků familiární adenomatózní polypózy?
Polypy tlustého střeva u familiární adenomatózní polypózy se začínají objevovat průměrně ve věku 15-16 let. U AFAP se vyskytují o něco později. Pokud vaši rodiče a lékař vědí, že jste ohroženi syndromem, pravděpodobně začnou provádět screening někdy po dovršení 10 let. Pokud to nevědí, mohou to být až vaše příznaky, které stav odhalí.
Příčiny – co je hlavní příčinou familiární adenomatózní polypózy?
Familiární adenomatózní polypóza je způsobena genovou mutací. Gen se nazývá APC. Tento gen má zabránit buňkám, aby se vymkly kontrole a tvořily nádory. Mutace zde ale zasahuje do funkce genu.
Genová mutace, která způsobuje familiární adenomatózní polypózu, je zárodečná mutace, což znamená, že k ní dochází během početí, nikoli během vašeho života. Pokud má mutaci jeden z vašich rodičů, máte 50% šanci, že ji zdědíte. Až 30 % těchto mutací je však původních, nikoli zděděných. To znamená, že asi 30 % pacientů s diagnózou familiární adenomatózní polypózy nebude mít rodinnou anamnézu onemocnění – tedy u 25–30 % pacientů jde o mutaci de novo.
Diagnóza – jak poznáte, že máte familiární adenomatózní polypózu?
Pokud chcete vědět, zda jste zdědili mutaci genu APC, můžete to zjistit pomocí genetického testování. Genetický test odebere vzorek DNA (jako je krev nebo sliny) a hledá specifické genové mutace. Pokud mutaci máte, dalším krokem je kolonoskopie ke kontrole adenomů.
Diagnóza familiární adenomatózní polypózy je založena na tom, že máte alespoň 100 polypů, nebo 20 pro AFAP, a mutaci APC.
Léčba – jaký je plán léčby u familiární adenomatózní polypózy?
Léčba familiární adenomatózní polypózy znamená celoživotní sledování a případně chirurgický zákrok. Na začátku, pokud máte méně polypů (nebo AFAP), lékař je může být schopen odstranit jednotlivě během screeningu kolonoskopie (tedy během polypektomie). Budete potřebovat operaci, když je jich příliš mnoho nebo se stanou rakovinnými.
Chirurgický zásah
Většina lidí s klasickou familiární adenomatózní polypózou podstoupí totální kolektomii (odstranění tlustého střeva). Váš lékař určí, kdy naplánovat operaci na základě vašich konkrétních rizikových faktorů. Bude s vámi také diskutovat o různých typech kolektomií, které byste mohli podstoupit.
Když si necháte odstranit tlusté střevo, vznikne mezera mezi tenkým střevem a konečníkem. Někdy může chirurg tyto konce znovu spojit dohromady. Pokud se to nedá, vytvoří stomii, nový otvor ve vašem břiše, aby vaše stolice takto vycházela ven.
Pravidelné screeningy
Lékař vám poradí, jak často si nechat udělat screeningové testy na různé typy nádorů na základě vašich osobních rizikových faktorů. U klasické familiární adenomatózní polypózy se často doporučuje roční kolonoskopie začínající ve věku 10 let a pokračující až do vaší kolektomie. U AFAP by roční screeningy měly začít ve věku 20 let.
Po kolektomii budete muset pokračovat s pravidelnými sigmoidoskopiemi, vyšetřeními konce vašeho GI (gastrointestinálního) traktu. Pokud máte konečník, měl by se kontrolovat každých 6 až 12 měsíců. Pokud byl váš konečník odstraněn, ale byl nahrazen ileálním vakem, měl by být kontrolován každý jeden až čtyři roky.
Další pravidelné kontroly mohou být:
- Horní endoskopie. Endoskop prochází krkem a do žaludku a dvanácterníku, aby tam lékař s jeho pomocí zkontroloval polypy. Pokud nějaké najde, lékař je může během procedury odstranit.
- Ultrazvuk pro screening rakoviny štítné žlázy nebo jater.
- CT (počítačová tomografie) nebo MRI (zobrazování magnetickou rezonancí) pro screening desmoidních nádorů.
Výhled a prognóza – jaká je očekávaná délka života někoho s familiární adenomatózní polypózou?
Sponzorováno
Bez včasné léčby je střední délka života 42 let. Ale s vhodnou péčí můžete žít normální život. Jakmile bude vaše tlusté střevo odstraněno, vaše největší riziko je z jiných gastrointestinálních rakovin nebo problematických desmoidních nádorů. Ty se ale vyskytují mnohem méně často než kolorektální karcinom.
Studie a zdroje článku
- Familial Adenomatous Polyposis Autoři: Gopal Menon; Stephanie Carr; Anup Kasi.
Autor článku
Líbil se vám náš článek? Sdílejte ho, uděláte nám radost
Štítky: Genetická onemocnění, Střeva
Přečtěte si také naše další články